Category: America
American Coronavirus News

Anthony Fauci canvasses a black neighbourhood to convince them to take the Covid-19 vaccine
It doesn’t go as planned.
Anthony Fauci strolls around an unknown Black neighborhood trying to convince residents that they should get vaccinated, claiming that it will protect their families from Covid-19 transmission if they have it. More...

Texas: H5N1 bird flu confirmed in a human
Texas has confirmed its first case of H5N1 avian flu in a human today. More...

Bill Maher nails the Covid pandemic
‘A lot of the dissenting opinions that were suppressed and ridiculed at the time have proven to be correct.’ More...

Highly Pathogenic Avian Influenza in Cattle in Texas, Kansas, New Mexico, Idaho and Michigan *14 updates*
The U.S. Government has announced that highly pathogenic avian influenza (HPAI) has been found in cattle in Texas and Kansas. More...
Covid-19 Rapid Antigen Tests Contain Highly Toxic Poison Sodium Azide
‘The vial in many rapid antigen kits includes the chemical Sodium Azide as a preservative agent. More...

U.S. Sen. Ron Johnson roundtable discussion: Contamination of mRNA vaccines with DNA
The latest Covid roundtable discussion, chaired by Senator Ron Johnson, featuring 21 guest speakers. More...

Third U.S. embalmer corroborates existence of white fibrous clots in cadavers
Another U.S. embalmer has given evidence to confirm the existence of white fibrous clots in cadavers. More...
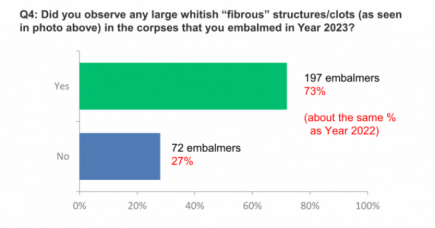
White fibrous clots found in at least 50% of all cadavers embalmed
Another interview by Dr John Campbell, and another Covid-19 bombshell. More...

Tucker Carlson interviews Dr Joseph Ladapo about DNA contamination of Covid-19 mRNA vaccines
The Tucker Carlson Network has released footage of an interview between Tucker Carlson and Dr Jospeh Ladapo, Surgeon General of Florida, exploring the subject of DNA contamination in mRNA vaccines. More...

Texas Attorney General sues Pfizer over Covid-19 vaccine claims and censorship
The Attorney General of Texas, Ken Paxton, is suing Pfizer for “misrepresenting COVID-19 vaccine efficacy and conspiring to censor public discourse.” More...