Category: P681H

France: new coronavirus variant B.1.640 detected in Finistère, Brittany
A new coronavirus variant has been identified in a school in Finistère in October 2021, and named B.1.640. More...

Preprint: Characterization of the emerging B.1.621 variant of interest of SARS-CoV-2
In this study, we reported the emergence and spread of the novel B.1.621 lineage of SARS-CoV-2, a new VOI with the insertion 146N and several amino acid substitutions in the Spike protein (T95I,Y144T, Y145S, R346K, E484K, N501Y and P681H). More...

Delta coronavirus variant now dominant in the USA
The highly transmissible Delta variant is the most common strain of the Covid-19 virus circulating in the U.S., More...

Russia: coronavirus revaccination planned for Moscow
Moscow Mayor Sergei Sobyanin has said that revaccination against coronavirus in the capital will take place with the Sputnik Light vaccine, which is also planned to be used to vaccinate labor migrants. More...

Russia: New “Moscow” coronavirus strain is Delta – Московский штамм коронавируса – Дельта
Russian newspapers are still referring to a new “Moscow” strain of Sars-CoV-2, however it seems that the strain may simply be Delta (B.1.617.2). More...
UK: Grant Shapps – “there’s a sort of Nepal mutation of the so-called Indian variant which has been detected and we just don’t know the potential for that to be a vaccine-defeating mutation”
Grant Shapps, UK Transport Secretary “we’ve seen two things really which caused concern. More...

UK: government risk assessment of coronavirus Delta variant B.1.617.2 – evidence of frequent vaccine failure or decreased effectiveness in humans * UPDATED *
- UPDATED IMAGE ABOVE
- * UK risk assessment for Delta variant as per 10th June 2021
Previous UK risk assessment as per 3rd June 2021

The UK government risk assessment for B.1.617.2 Delta variant has been published today and it’s NOT good news. More...

Preprint: B.1.617 coronavirus variants show enhanced spike cleavage by furin
“The spike (S) glycoprotein of the SARS-CoV-2 virus that emerged in 2019 contained a suboptimal furin cleavage site at the S1/S2 junction with the sequence 681 PRRAR/S 686. More...
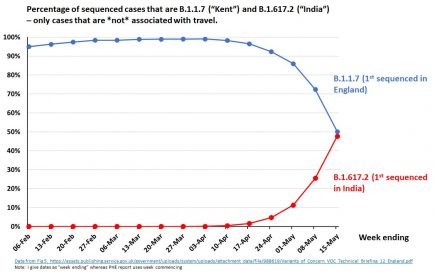
UK: PHE data shows B.1.617.2 now outcompetes B.1.1.7
More amazing work by Prof. Christina Pagel in deciphering the latest Public Health England data dumps for B16172. More...
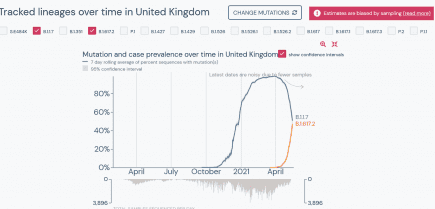
UK: coronavirus variant B.1.617.2 within hours of outcompeting B.1.1.7 in Britain
Well that escalated quickly. It took just a little over one month to do it, but B.1.617.2 is now just hours away from outcompeting B.1.1.7 (aka the UK or Kent variant) on a national scale in Great Britain. More...