Category: P.1

WHO: The end of the pandemic is plausible
The Covid-19 pandemic has entered a new phase with the Omicron variant, which could infect 60 percent of people in Europe by March, and could bring it to an end, the WHO Europe director said Sunday. More...

Vaccine breakthrough cases in Washington State to 22nd September 2021
This week’s update from Washington State showing vaccine breakthrough cases and which Sars-CoV-2 variant is causing them up to 22nd September 2021. More...
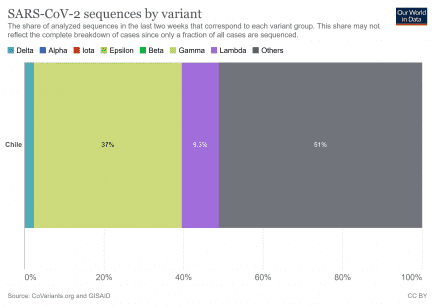
The stunning rise of the Mu variant
The new Mu variant, B.1.621, detected for the first time in Colombia, surpasses Lambda and Delta and is already ranked as the second with the highest circulation in Chile. More...

Vaccine breakthrough cases in Washington State to 1st September 2021
This week’s update from Washington State showing vaccine breakthrough cases and which Sars-CoV-2 variant is causing them up to 1st September 2021. More...

Vaccine breakthrough cases in Washington State to 25th August 2021
This week’s update from Washington State showing vaccine breakthrough cases and which Sars-CoV-2 variant is causing them up to 25th August 2021. More...

UK: Vaccine breakthrough rate for Delta infections still increasing
The latest UK PHE Technical Briefing has just been published, and it shows that the vaccine breakthrough rate for Delta infections in the UK continues to increase by about 6% per month. More...

280 cases of Delta variant with N501Y, some with E484K too
Delta + Alpha + Beta + Gamma recombinant variant found in Turkey. More...

USA: 55% increase in Delta vaccine breakthrough cases in Washington State
This week’s update from Washington State showing vaccine breakthrough cases and which Sars-CoV-2 variant is causing them up to 11th August 2021. More...

USA: Washington State Vaccine Breakthroughs by Variant up to 28th July 2021
This week’s update from Washington State showing vaccine breakthrough cases and which Sars-CoV-2 variant is causing them up to 28th July 2021. More...

USA: Washington State Vaccine Breakthroughs by Variant up to 21st July 2021
This week’s update from Washington State showing vaccine breakthrough cases and which Sars-CoV-2 variant is causing them. More...