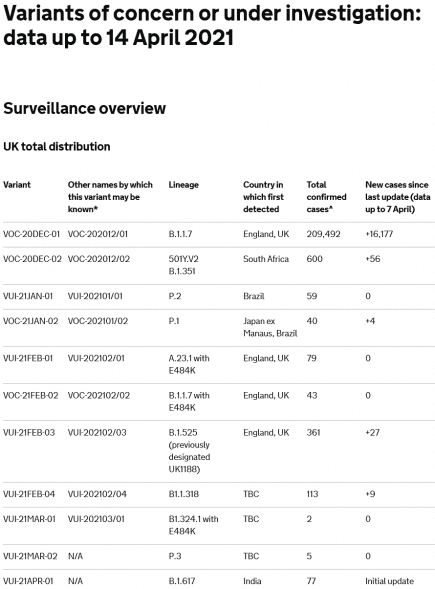
Category: N501Y

France: new Sars-CoV-2 variant B.1.640.2 with N501Y and E484K detected
Twelve SARS-CoV-2 positive patients living in southeastern France are infected with a variant with atypical mutations. More...

France: new coronavirus variant B.1.640 detected in Finistère, Brittany
A new coronavirus variant has been identified in a school in Finistère in October 2021, and named B.1.640. More...
Delta 4+ variant: Complete vaccine escape & enhanced infectivity with Pfizer vaccine
“Here we suggest an evolutionary pathway by which the Delta variant could achieve complete escape from vaccine-induced immunity”. More...

280 cases of Delta variant with N501Y, some with E484K too
Delta + Alpha + Beta + Gamma recombinant variant found in Turkey. More...

Preprint: Characterization of the emerging B.1.621 variant of interest of SARS-CoV-2
In this study, we reported the emergence and spread of the novel B.1.621 lineage of SARS-CoV-2, a new VOI with the insertion 146N and several amino acid substitutions in the Spike protein (T95I,Y144T, Y145S, R346K, E484K, N501Y and P681H). More...
Preprint: Persistent coronavirus infection and intra-host evolution in association with advanced HIV infection
“Here we present a case of prolonged infection of greater than 6 months with shedding of high titer SARS-CoV-2 in an individual with advanced HIV and antiretroviral treatment failure. More...

France: 46 cases of B.1.1.7 with E484Q mutation in Bordeaux coronavirus outbreak of VOC 20I/484Q
The French city of Bordeaux is to fast-track vaccinations for residents in one neighbourhood, opening access the jab for all adults after nearly 50 people tested positive for a “very rare” variant of Covid-19.
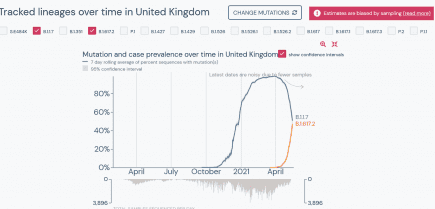
UK: coronavirus variant B.1.617.2 within hours of outcompeting B.1.1.7 in Britain
Well that escalated quickly. It took just a little over one month to do it, but B.1.617.2 is now just hours away from outcompeting B.1.1.7 (aka the UK or Kent variant) on a national scale in Great Britain. More...

UK SAGE: Lab studies suggest that the N501Y spike protein mutation increases binding to rat and mouse ACE2 leading to viral replication
A report issued by the UK government’s Scientific Advisory Group for Emergencies (SAGE) found the likelihood that a variant of concern (VOC) that has arisen in humans could infect a rodent and then spread among the animals is high. More...