Category: E484K

China: Swans infected with SARS-CoV-2
SARS-CoV-2 positive sequences from two SWANS have been deposited by Chinese scientists on Gisaid. More...

France: new Sars-CoV-2 variant B.1.640.2 with N501Y and E484K detected
Twelve SARS-CoV-2 positive patients living in southeastern France are infected with a variant with atypical mutations. More...
Delta 4+ variant: Complete vaccine escape & enhanced infectivity with Pfizer vaccine
“Here we suggest an evolutionary pathway by which the Delta variant could achieve complete escape from vaccine-induced immunity”. More...

280 cases of Delta variant with N501Y, some with E484K too
Delta + Alpha + Beta + Gamma recombinant variant found in Turkey. More...

Preprint: Characterization of the emerging B.1.621 variant of interest of SARS-CoV-2
In this study, we reported the emergence and spread of the novel B.1.621 lineage of SARS-CoV-2, a new VOI with the insertion 146N and several amino acid substitutions in the Spike protein (T95I,Y144T, Y145S, R346K, E484K, N501Y and P681H). More...
Preprint: Persistent coronavirus infection and intra-host evolution in association with advanced HIV infection
“Here we present a case of prolonged infection of greater than 6 months with shedding of high titer SARS-CoV-2 in an individual with advanced HIV and antiretroviral treatment failure. More...
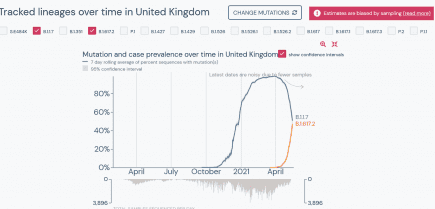
UK: coronavirus variant B.1.617.2 within hours of outcompeting B.1.1.7 in Britain
Well that escalated quickly. It took just a little over one month to do it, but B.1.617.2 is now just hours away from outcompeting B.1.1.7 (aka the UK or Kent variant) on a national scale in Great Britain. More...

UK: Detection of Spike Mutations; D80G, T95I, G142D, △144, N439K, E484K, P681H, I1130V, and D1139H, in B.1.1.482 Lineage (AV.1) Samples from South Yorkshire, UK
“Here we report the discovery of 24 recent B.1.1.482 pango lineage SARS-CoV-2 viruses in South Yorkshire that have acquired approximately 23 additional mutations when compared to the majority of other samples of this lineage. More...
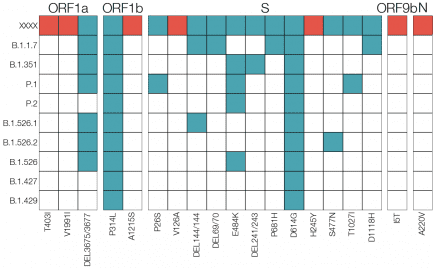
Europe: B.1.620 coronavirus variant – definitely one to watch
“Last month, Gytis Dudas was tracking a concerning new coronavirus variant that had triggered an outbreak of COVID-19 in his native Lithuania and appeared sporadically elsewhere in Europe and in the United States. More...

Cameroon: travel-driven emergence and spread of SARS-CoV-2 lineage B.1.620 with multiple VOC-like mutations and deletions in Europe
“In this study we have presented evidence that a SARS-CoV-2 lineage designated B.1.620, first detected in Europe in late February, is associated with Central Africa, where it appears to circulate at very high prevalence, and has been introduced into Europe and the US on multiple occasions. More...