Category: L452R

Preprint: L452R mutation increases Omicron variant fusogenicity and infectivity
Here, we developed an L452R mutated Omicron variant (Omicron-L452R) and found that the Omicron-L452R variant rescued fusogenicity and strengthened the high infectivity by enhancing the cleavage of the spike protein. More...

280 cases of Delta variant with N501Y, some with E484K too
Delta + Alpha + Beta + Gamma recombinant variant found in Turkey. More...
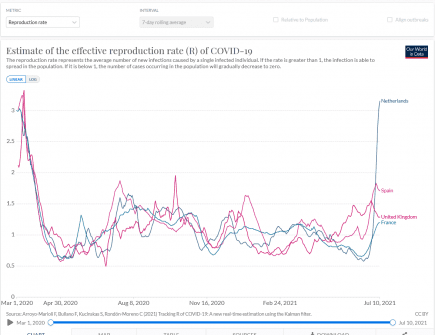
Netherlands: the reproduction R rate is now 2.17 – the highest since February 2020
For the first time since the start of the coronavirus pandemic, people in the Netherlands infected with the SARS-CoV-2 virus were likely infecting at least two other people. More...

India: Delta Plus Delta-AY.1 dominant in Tripura – found in 138 out of 151 coronavirus sequences
At least 138 of the 151 Covid-19 samples sent by Tripura for genome sequencing have tested positive for the Delta Plus variant, State Health Surveillance Officer Dr Deep Kumar Debbarma said. More...
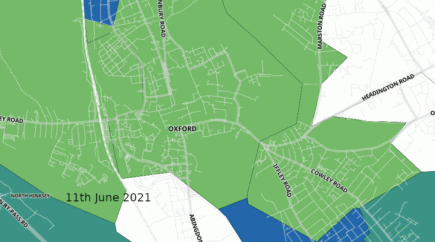
UK: Oxford overwhelmed by Delta coronavirus variant in just 3 weeks
Covid cases in Oxford have increased fivefold in one week, almost entirely among 15 to 24-year-olds, according to the latest official figures. More...

Israel: 56% of serious coronavirus cases in hospitals are fully vaccinated
Eran Segal, a COVID expert and one of the top government advisers to the Israeli coronavirus cabinet said that while 56% of current serious COVID cases occur among fully-vaccinated individuals, the vaccine nevertheless remains the best possible protection against the disease. More...
Preprint: Transmission event of Delta coronavirus variant reveals multiple vaccine breakthrough infections
Here we describe a transmission of a Delta variant containing SARS-CoV-2 strain, between family members associated with events surrounding a wedding with 92 attendees, near Houston, Texas. More...

USA: Delta now the dominant variant in California
California Department of Public Health say 35.6% of coronavirus variants sequenced in June have been identified as Delta variant. More...

Delta coronavirus variant now dominant in the USA
The highly transmissible Delta variant is the most common strain of the Covid-19 virus circulating in the U.S., More...

Poland: 106 cases of Delta and 12 cases of Delta Plus aka Delta-AY.1 coronavirus variant
Poland has confirmed 106 cases of the Delta and 12 cases of the Delta Plus (Delta-AY.1 or Delta with K417N mutation) coronavirus variants that originated in India, a deputy health minister has announced. More...