Category: P.2

USA: Washington State Vaccine Breakthroughs by Variant up to 21st July 2021
This week’s update from Washington State showing vaccine breakthrough cases and which Sars-CoV-2 variant is causing them. More...
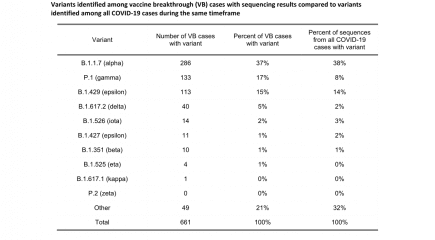
USA: Vaccine breakthough cases in Washington State by coronavirus variant to 30th June 2021
Washington State has recently published an update on the number of vaccine breakthrough cases. More...
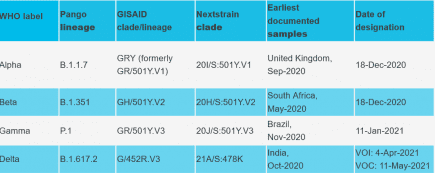
WHO announce new names for Sars-CoV-2 coronavirus variants
The WHO have announced new names for the confusingly named sars-cov-2 variants. More...

Brazil: new coronavirus strain P.1.2 confirmed circulating in Rio de Janeiro
The State Department of Health of Rio de Janeiro (SES-RJ) announced the identification of the new variant of the Covid virus in Rio de Janeiro late this morning. More...

Brazil: P.1 mutates into new coronavirus variant P.1.2
A new variant of the Brazilian coronavirus has been identified this week, according to Rio de Janeiro health authorities. More...

Brazil: First confirmed death from coronavirus REINFECTION reported
A 39-year-old Brazilian man who died of COVID-19 last month was suffering from a second bout of the illness, researchers said on Tuesday, making it the country’s first confirmed death from coronavirus reinfection. More...
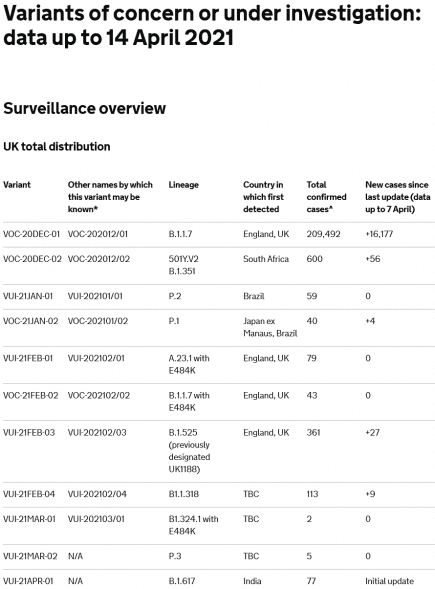

Brazil: rapid spread and high impact of the coronavirus VOC P.1 in Sao Paulo – March 2021
Here, we show evidence of how fast the VOC P.1 has spread in the most populated city in South America – Sao Paulo. More...

Genetic Evidence and Host Immune Response in Persons Reinfected with SARS-CoV-2 coronavirus in Brazil
A paper to be published in May in the U.S. More...
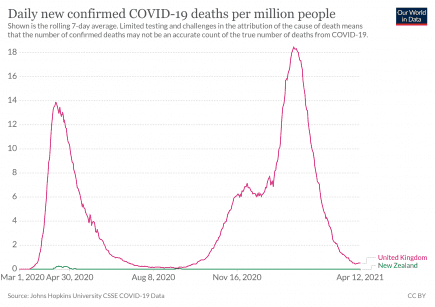
Boris Johnson: “many more people will lose loved ones to coronavirus”
12th March 2020 – Boris Johnson: “many more people will lose loved ones to coronavirus” (Guardian). More...