Category: Brazil

Brazil: First case of monkeypox in a dog
The Brazilian Ministry of Health has been notified of the first confirmed case of monkeypox in a domestic animal. More...

Brazil: 2 cases of XQ Omicron recombinant reported
Two cases of the subvariant of Covid-19 called Omicron XQ were registered in São Paulo this Thursday, 5th May 2022. More...

SARS-CoV-2 potential recombinants in Brazil, Belgium, France, Netherlands, US, Finland, Puerto Rico, Spain
SARS_CoV-2 recombinants seem to have been popping up across the globe recently. More...

Research: Evidence of SARS-CoV-2 before Wuhan outbreak in 2019
In the USA, SARS-CoV-2 reactive antibodies were detected in over 100 blood samples collected in several different states in early December 2019. More...

Brazil: previously infected and fully vaccinated subjects with high anti-S IgG titers are susceptible to infection by VOC
Here we show that even previously infected and fully vaccinated subjects with high anti-S IgG titers are susceptible to infection by VOC, highlighting that immune responses induced by either natural infection or vaccination may not be sufficient to prevent infection by SARS-CoV-2 variants. More...

Brazil: new strain of coronavirus identified in Rio de Janeiro, designated P5
Monitoring in Rio de Janeiro, Brazil, has identified a possible new Sars-Cov-2 lineage, originating from P.1.1.28, in the region of Barra Mansa and Porto Real, on the border with the State of São Paulo, and which has only now been called P5. More...

Brazil: new strain of coronavirus with L452R mutation found in São Paulo interior named P.4
The Brazilian Society of Virology (SBV) yesterday, May 25, confirmed the identification of a new Brazilian coronavirus strain, denominated P4. More...
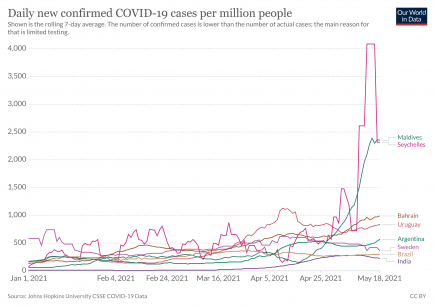
The highest ever coronavirus daily case rates recorded on Planet Earth – and it’s not India or Brazil
The highest ever daily country case figure for coronavirus on Planet Earth was recorded in The Seychelles in May 2021. More...

Brazil: new coronavirus strain P.1.2 confirmed circulating in Rio de Janeiro
The State Department of Health of Rio de Janeiro (SES-RJ) announced the identification of the new variant of the Covid virus in Rio de Janeiro late this morning. More...

Brazil: P.1 mutates into new coronavirus variant P.1.2
A new variant of the Brazilian coronavirus has been identified this week, according to Rio de Janeiro health authorities. More...